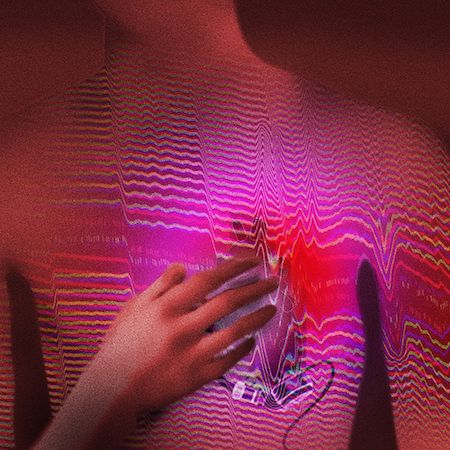
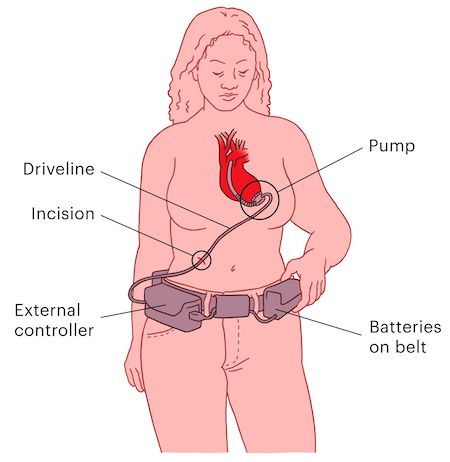
Inspectors repeatedly found manufacturing and device quality problems with the HeartWare heart pump. But the FDA did not penalize the company, and patients had the device implanted on their hearts without knowing the facts. John Winkler II was dying of heart failure when doctors came to his hospital bedside, offering a chance to prolong his life. The HeartWare Ventricular Assist Device, or HVAD, could be implanted in Winkler’s chest until a transplant was possible. The heart pump came with disclaimers of risk, but Winkler wanted to fight for time. He was only 46 and had a loving wife and four children, and his second grandchild was on the way. So, in August 2014, Winkler had surgery to implant the device. A golf-ball-sized rotor was attached to his left ventricle to pump blood through a tube and into his aorta. A cable threading out of a small incision in his waist connected to a battery-powered controller strapped to his body. If something went wrong, an alarm as loud as a fire drill would sound. Winkler returned home weeks later and, as he regained his strength, became hopeful about the future. He started making plans to visit colleges with his daughter, and was able to host his parents and new grandchild for Christmas. “He was doing so much better,” his wife, Tina Winkler, said. “We thought he was coasting until he got his transplant.” What John Winkler didn’t know: Months before his implant, the Food and Drug Administration put HeartWare on notice for not properly monitoring or repairing HVAD defects, such as faulty batteries and short circuits caused by static electricity, that had killed patients. The agency issued a warning letter, one of its most serious citations. It demanded fixes within 15 days, but took no decisive action as problems persisted. Ten days after Christmas 2014, Winkler’s two teenage children heard the HVAD’s piercing alarm and ran upstairs. They found their father collapsed on his bedroom floor, completely unresponsive. Kelly, 17, dropped to his side and tried to copy how people on television did CPR. She told her brother to call 911, and over the device’s siren did her best to hear instructions from the operator. When paramedics arrived and assessed her father, one made a passing comment that has haunted Kelly ever since: “Well, his toes are already cold.” He died two days later. Medtronic, the company that acquired HeartWare in 2016, settled a lawsuit by the family last year, admitting no fault. Tina Winkler believes her children blamed themselves for their father’s death. “Those two kids have never been the same,” she said. “I think they feel like they didn’t do things they needed to do.” But it was the FDA that failed to protect Winkler and thousands of other patients whose survival depended on the HVAD, a ProPublica investigation found. As HeartWare and Medtronic failed inspection after inspection and reports of device-related deaths piled up, the FDA relied on the device makers to fix the problems voluntarily rather than compelling them to do so. The HVAD was implanted into more than 19,000 patients, the majority of whom got it after the FDA found in 2014 that the device didn’t meet federal standards. By the end of last year, the agency had received more than 3,000 reports of patient deaths that may have been caused or contributed to by the device. Among them were reports of deaths the company linked to serious device problems: a patient who vomited blood as a family member struggled to restart a defective HVAD; a patient who bled out internally and died after implant surgery because a tube attached to the pump tore open; a patient whose heart tissue was left charred after an HVAD short-circuited and voltage surged through the pump. The ineffective regulatory oversight of the HVAD is emblematic of larger, more systemic weaknesses. For decades, the FDA and its Center for Devices and Radiological Health have been responsible for ensuring that high-risk medical devices are safe and effective. Yet they mostly rely on manufacturers to identify and correct problems. The agency says it can seize products, order injunctions against companies or issue fines, but it rarely does so, preferring instead for companies to make fixes voluntarily. When federal investigators found repeated manufacturing issues with the HVAD for years, the FDA didn’t penalize the company, even as the company issued 15 serious recalls of the device starting in 2014, the most of any single high-risk device in the FDA’s database. Thousands of patients with recalled models needed to have external HVAD parts replaced or take extra caution while handling their devices and monitor them for signs of malfunctions that could cause injury or death. Meanwhile, the processes to inform the public through formal FDA notices and messages to healthcare providers repeatedly failed and left patients in the dark about known problems with the HVAD. “Patients have no idea, and they rely on the FDA to ensure the safety and effectiveness of high-risk devices,” said Dr. Rita Redberg, a cardiologist at the University of California, San Francisco who studies medical device regulation. “How can you not take action on a warning letter with these serious issues with very sick patients?” In response to ProPublica’s findings, the FDA said it had been closely monitoring issues with the HVAD. It said that after Medtronic acquired HeartWare in 2016, it met with the company more than 100 times to ensure problems were being fixed and to review safety concerns related to the heart pump. The agency also said it initiated formal reviews of new device modifications and continually tracked whether the HVAD had a “reasonable assurance of safety and effectiveness.” “Our decisions that we made along the way have always been patient-focused,” said Dr. William Maisel, the director of product evaluation and quality at the FDA’s device division. He added that more than 80% of companies fix their problems by the time the FDA reinspects. That did not happen with the HVAD. In 2016 and 2018, inspectors found that issues detailed in the 2014 warning letter remained unresolved. Medtronic told the FDA last year that it had fixed the problems, but, before the agency could verify the claim, inspections were paused because of the coronavirus pandemic. In June, Medtronic stopped HVAD sales and implants. The company conceded that a competing device was safer after a new study showed the HVAD had higher rates of death and neurological injury. Medtronic also cited a 12-year-old problem with its devices not restarting if they disconnect from power, leaving patients’ hearts without support. Medtronic declined to make CEO Geoffrey Martha or president of mechanical heart support Nnamdi Njoku available for interviews. In an email, a spokesperson said, “There is nothing more important to Medtronic than the safety and well-being of patients.” The email continued, “Medtronic takes this matter very seriously and, over the past five years, we have worked closely with FDA and engaged external experts to resolve the issues noted in the warning letter. FDA is aware of the steps Medtronic has taken to address the underlying concerns.” The company said it will have a support system in place for the 4,000 patients worldwide and 2,000 in the United States who still rely on the HVAD. Medtronic will station 20 specialists across the globe to help with device maintenance and patient education. A centralized engineering team will also provide technical support and troubleshooting for patients and medical staff. Medtronic said it will also offer financial assistance if insurance doesn’t fully cover the surgery to replace a device with a competing product, but only if a doctor decides it’s medically necessary. Patients with HVADs have little choice but to hope the devices keep working: The surgery to remove HVADs is so risky that both Medtronic and the FDA advise against it. The device is meant to be left in place until its wearer gets a heart transplant. Or dies.
Warning Signs In late 2012, HeartWare, then an independent company headquartered in Massachusetts, won FDA approval to sell a new device that could keep heart failure patients alive and mobile while awaiting a transplant. A competing device, the HeartMate, was already gaining attention, with high-profile patients like former Vice President Dick Cheney, a heart attack survivor who eventually got a transplant after using the device for 20 months. The HVAD offered a smaller option that could even be used in children, and it led to a string of publicized successes. A fitness model was able to return to the gym. A 13-year-old with heart defects could attend school again. Medtronic’s YouTube page features 16 interviews with grateful patients and families. The patients who received HVADs had already been in grave peril. They had advanced heart failure, serious enough to need blood pumped out of their hearts artificially. Most patients were older than 50, but there were also younger patients with heart defects or other cardiac conditions. The device provided help but brought its own risks. Implanting it required invasive open-heart surgery, and clots could develop inside the pump, which, in the worst cases, led to deadly strokes. The device also came with a steep price tag. Each HVAD cost about $80,000, and, even though HeartWare never made a profit as an independent company, in 2015 device sales brought in $276 million in revenue. For many severe heart failure patients, the opportunity to survive longer and return to normal life made the device worth the risks and cost. But patients were unaware the FDA started finding manufacturing issues at HeartWare’s Miami Lakes, Florida, plant as early as 2011, when the device was still seeking approval. Among the findings, a federal inspector expressed concerns that engineering staff “were not completely reviewing documents before approving them” and found one employee assigned to monitoring device quality had missed several required monthly trainings. HeartWare leadership promised quick corrective action, according to FDA documents. Then, in 2014, the FDA found more serious lapses, detailed in federal inspection reports. For example, HeartWare knew of 119 instances in which batteries failed unexpectedly, which could leave the pump powerless, stopping support for the patient’s heart. But the company didn’t test the batteries in inventory for defects, or the batteries of current patients, even though one person’s death had already been linked to battery failure. The company also received complaints that static electricity could short-circuit its devices. It learned of at least 27 such cases between 2010 and 2013, including four that resulted in serious injuries and two that led to death. HVAD patients would need to avoid contact with certain household objects like televisions or vacuum cleaners — anything that could create strong static electricity. HeartWare added warnings to the patient manual and redesigned its shield to protect the device controller, but the FDA found that the company didn’t replace shields for devices already being used by current patients or produced and sitting in inventory. Continuing quality control concerns led to the FDA warning letter in June 2014. The document labeled the HVAD as “adulterated,” meaning the device did not meet federal manufacturing standards. The agency gave HeartWare 15 days to correct the problems or face regulatory action. Still, investment analysts who followed HeartWare believed the warning posed little risk to the company’s business prospects. One described it as being “as benign as possible.” The 15-day deadline passed, and the FDA never penalized the company.
The agency told ProPublica it had provided additional time because HeartWare was a relatively new manufacturer and the HVAD was a complicated device. It also said it avoided punitive action to make sure patients with severe heart failure had access to this treatment option. “We’re talking about the sickest of the sick patients who really have very few alternatives,” Maisel, the head of device quality, said. But the HeartMate, the competing device, was available and already being used by the majority of patients. When Medtronic stopped HVAD sales, both companies said the HeartMate could fill the gap. Inspectors continued to find problems at HeartWare facilities in 2015, 2016, 2017 and 2018. In the most recent report in 2018, inspectors identified seven separate violations at the HVAD plant, including three previously cited in the 2014 warning letter. The company was still mishandling newly discovered defects like pins connecting the controller to a power source that could bend and become unusable, and controllers built with incompatible parts that could chemically react and “attack” the plastic exterior. Again, the inspection report said the company “promised to correct” the issues. “What penalty is there for noncompliance? There isn’t one,” said Madris Kinard, a former public health analyst with the FDA and the CEO of Device Events, a software company that analyzes FDA device data. “There’s nothing the FDA is doing that penalizes, in any true sense of the matter, the manufacturer.” By the time sales were halted last month, the HVAD had become the subject of 15 company-initiated “Class I” recalls for dangerous device problems that could cause injury or death. One recall came with a warning sent to health care providers in December that said pumps were failing to start up properly. The pattern of malfunctions was almost as old as the device itself, the company later admitted when it halted device sales in June. But even recent patients were completely unaware of the problem. “A No-Brainer” When children asked Latoya Johnson Keelen about the cable that came out of her side and connected to a controller on her hip, she told them she was Iron Woman. For a while, she felt invulnerable with the HVAD on her heart. Johnson Keelen, who lives in the Atlanta suburbs, learned she needed the device after delivering her fourth child, Isaiah, in early 2018. Doctors diagnosed her with postpartum cardiomyopathy, a rare and mysterious form of heart failure that afflicts mothers during pregnancy or after birth. Black mothers in the South have among the highest rates of the illness. Some mothers quickly regain heart function, some only partially recuperate and others never recover. Tests showed that Johnson Keelen, then 42, was suddenly in end-stage heart failure. Her body’s immune response at the time was too strong for her to receive a heart transplant. Doctors gave her two choices: an HVAD or end-of-life hospice care. “It became a no-brainer,” she said. “I just had a baby. I just gave birth. I’m not ready to plan for a funeral.” Johnson Keelen, a woman of faith, believed God would heal her, either through a medical advancement or a miracle. She thought the HVAD was the answer.
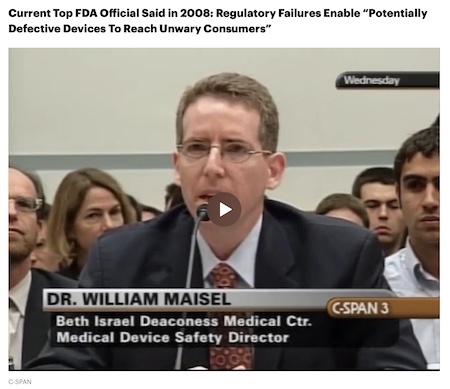
Living with a life-sustaining medical device was difficult at first for the fiercely independent mother. She had to leave her job as a public health communications specialist, ask her older sons to change her bandages and lean heavily on her new husband, only a year into their marriage. But, for about three years, she found comfort in the soft humming of the HVAD’s spinning rotor at night. It served as a lullaby for her new baby when he lay on her chest. She said she was never told about the manufacturing problems the FDA repeatedly found at HeartWare’s facilities or about device recalls, including one sent to patients in December 2020. The notice said the device sometimes wouldn’t restart properly, which had led to two patient deaths at that point. It warned that current patients should always keep at least one power source, a battery or an AC or DC adapter, connected at all times to avoid the need for a restart. Two months after that notice, Johnson Keelen was getting her kids ready for school when the HVAD’s low-battery alarm blared. She had unplugged the battery to replace it without realizing her wall adapter was disconnected. Once before, Johnson Keelen had simply plugged the charger back into the outlet and her device restarted. But this time it wouldn’t. As an emergency alarm sounded, she called the ventricular-assist team assigned to her case, and a specialist directed her to switch out the device controller. Nothing changed, and panic crept into the voice on the phone. An ambulance took Johnson Keelen to a hospital where medical staff used several backup controllers to try to start the pump. Still nothing. Doctors and nurses tried to keep calm, but Johnson Keelen could see fear and shock on their faces. Without the HVAD, her only options were a transplant or a completely new pump. Doctors scurried to locate a donor heart and airlifted her for an emergency transplant. But while running tests, the medical team was stunned to find that Johnson Keelen’s miracle had occurred: Her heart was once again pumping blood on its own. She had a new choice. She could avoid the risks of transplant rejection and open heart surgery during the pandemic by leaving the device on her functioning heart, while cutting the wires, removing the external components and sealing the pump. She chose to trust her newly functioning heart, and leave the decommissioned HVAD inside her. Three months later, when Medtronic said it was stopping HeartWare sales and implants, its announcement cited the problem with pumps not restarting among the reasons. Company-Led Oversight If evidence suggests a medical device may be linked to a serious patient injury or death, hospitals and other health care facilities must submit a report to the manufacturer and the FDA. Device companies must also submit reports if they learn independently of any incidents. By the end of 2020, roughly 3,000 death reports and 20,000 injury reports related to the HVAD had been filed with the FDA. Any details that could identify patients, like their age or gender, are removed from the publicly available reports. Most only have limited details about circumstances surrounding deaths or injuries. But it’s clear from the reports on the HVAD that some of these outcomes could be linked to problems previously identified by FDA inspectors. Doctors attempted CPR for two hours after an electrostatic shock short-circuited one patient’s device in 2014, a few months after the FDA inspection that year. An autopsy revealed voltage had caused “deep charring” of the tissue inside the patient’s chest. Friends found another patient dead in the kitchen, with groceries still on the counter, in 2018 after their device, which did not have the recommended static shield, short-circuited. Last year, paramedics found a patient with the device disconnected from power. They struggled to restart the device, but it wouldn’t plug back into the power source because the connector pins were bent. The patient would die at the hospital. In most cases, the FDA turned to the company to investigate whether a malfunction caused or contributed to the incidents. But the FDA has long known HeartWare and Medtronic could not be relied on to properly submit HVAD incident reports. In 2014, the FDA cited HeartWare because in at least 10 cases, there were no documents showing the company attempted to investigate. In 2016, the agency wrote another citation when the company was late in reporting more than 200 cases, some more than a year past their 30-day reporting deadlines, and failed to report malfunctions that occurred during clinical trials. The FDA told ProPublica the agency increased its monitoring of HVAD reports, and Medtronic hired new employees to submit timely reports. But by 2018, its backlog had only grown, with 677 late case filings. Again, the FDA did nothing beyond telling the company to fix the problem and further increasing its monitoring. In an email, Medtronic said it “has robust systems in place to monitor the safety of all of our products, including the HVAD device.” The email said, “When any potential safety issues are identified, those issues are thoroughly investigated and relevant information is shared with regulators and healthcare providers.” The company didn’t respond to the pattern of late reports and incomplete investigations identified in FDA inspections. Maisel, the director of FDA device evaluation and quality, once criticized asking companies to investigate their own devices. In 2008, as a practicing cardiologist, he testified to the U.S. House oversight committee about his concerns. “In the majority of cases, FDA relies on industry to identify, correct and report the problems,” he said. “But there is obviously an inherent financial conflict of interest for the manufacturers, sometimes measured in billions of dollars.”
Maisel has since had a change of heart. When asked about his 2008 testimony, he told ProPublica that he now believes the regulatory system “generally serves patients well” and “most companies are well intentioned.” HeartWare’s track record of questionable investigations was glaring in John Winkler II’s case. A report submitted by HeartWare that matches the dates and details of Winkler’s case shows the company decided there was “no indication of any device malfunctions.” It told the FDA that the device couldn’t be removed from the body because the hospital said his family declined an autopsy. HeartWare added that the evidence of the device’s role in Winkler’s death was inconclusive. Yet little of this appears to be true. Documents reviewed by ProPublica show an autopsy of the heart and lungs was performed a day after the death. Tina Winkler said she was told the pump was removed from her husband’s body and was available for inspection. A year after John Winkler’s death, HeartWare recalled 18,000 potentially faulty batteries produced between 2013 and 2015. Tina Winkler came across the notice online and found her husband’s battery serial numbers on the list. The company never contacted her about it or any further investigation, she said. Rewards, Not Penalties As deaths and recalls mounted, HeartWare and Medtronic touted additional FDA approval to treat more patients and their attempts to develop new cutting-edge devices. With the company on notice under the 2014 warning letter, HeartWare geared up to begin human trials on a smaller heart pump, called the MVAD or Miniaturized Ventricular Assist Device. It would be powered by a new algorithm to more efficiently pump blood. Industry analysts predicted robust sales. In July 2015, implantations were set to begin on a select group of 60 patients in Europe and Australia. But they were abruptly stopped less than two months later after only 11 implants. Patients experienced numerous adverse events, including major bleeding, infection and device malfunction, according to published data.
HeartWare’s stock price plummeted from about $85 to $35 by October 2015. The next year, Medtronic bought HeartWare for $1.1 billion, replacing much of the company’s leadership shortly after. Some former HeartWare investors filed a class action lawsuit in January 2016 alleging deception in the development of the MVAD. According to the accounts of six anonymous former employees in the lawsuit, the details mirror the scandal surrounding Theranos, the former blood test company charged with fraud for raising more than $700 million by allegedly lying about its technology. Where Theranos made empty promises of a test that only needed a few drops of blood, the suit alleges HeartWare promoted a life-sustaining medical device that former employees said had many problems and actually worsened blood flow, increasing clotting risks. “Nothing really worked right,” one former HeartWare manager said in the lawsuit, citing “improper alarms, improper touch screen performance, gibberish on display screens — just so many alerts and problems.” Leadership proceeded with human testing anyway, the suit alleges. Months later, at an investor conference, HeartWare leadership acknowledged the pump and algorithm led to multiple adverse events. For two patients in particular, the algorithm would direct the pump to speed up so fast that it would try to suck up more blood than was available inside the heart for prolonged periods of time. HeartWare and Medtronic settled the investor suit for $54.5 million in 2018, admitting no fault. None of the allegations slowed the FDA as it gave Medtronic additional approval and support for its heart pump technologies. In September 2017, the agency approved the HVAD as “destination therapy” for patients who were not heart transplant candidates and would rely on the device for the rest of their lives. “We’re really excited about our HVAD destination therapy approval,” a Medtronic executive said on an investor earnings call. “That’s a real game changer for us in that market.” Two years later, Medtronic announced it was developing a fully implantable version of the HVAD that would no longer need a cable coming through the waist to connect to power. Even though issues with the HeartWare device had been unresolved for five years at that point, the FDA accepted the pitch into its new fast-track approval process for high-risk devices. “Slipped Through The Cracks” After Johnson Keelen’s pump failed in February, she found a news story about the recall notice sent to medical providers two months prior. It said the company had identified a problem with pump restarts that could cause heart attacks or serious patient harm. Nineteen patients had been seriously injured so far, and two people had died. The recall warned that patients should be careful to avoid disconnecting the device’s power sources. “I kept seeing Medtronic on record saying they notified patients,” Johnson Keelen said. “Who did they contact? No one told me.” Her doctor later told her she must have “slipped through the cracks,” she said.
The current system for informing patients of new safety concerns with high-risk devices relies on a communication chain that can easily break. The device company contacts the FDA and health care providers that work with device patients. The FDA typically issues a public notice, while health professionals contact their patients. But the agency admits most patients don’t know to look for formal FDA postings. And, experts say, the medical system can lose track of who needs to be notified, especially if a patient moves or switches primary care physicians. Tina Winkler still wonders why she was never told about FDA-known safety issues with the HVAD. She said her husband’s medical team “had to teach me how to clean his wound, how to change his batteries and what to do if alarms go off. And they never mentioned any of this.” She said, “If we had all the facts, there’s no way he would have gotten that device implanted in his heart.” When FDA inspectors find serious safety issues with a medical device, inspection reports are not posted online or sent to patients. The public can obtain reports through a Freedom of Information Act request, but the agency’s records department has said new requests can be stuck behind a year-long backlog. Patients can find warning letters online in a searchable database of thousands of letters from different FDA divisions, including the center for devices. But HeartWare’s 2014 letter is no longer available for public review because the website purges letters older than five years. There are also few documents available in state courts about faulty products, because of restrictions on lawsuits related to medical devices. The restrictions date back to a 2008 Supreme Court decision in a case against Medtronic. The court found that U.S. law bars patients and their survivors from suing device makers in state court, essentially because their products go through such a rigorous FDA approval process. Two recent patient lawsuits against HeartWare and Medtronic, including one filed by Tina Winkler, were moved from state court to federal court. In both cases, Medtronic filed to dismiss the cases because of the U.S. law that protects device companies. Medtronic and the families reached private settlements soon after. Winkler and an attorney for the other family said they could not comment on their settlements. Johnson Keelen, with a decommissioned HVAD still attached to her heart, wonders what that means for her and other patients’ chances of recourse. “Why isn’t anyone now stepping up for the patient?” she asked. “They are now liable for taking care of us because we relied on them.” “Run Its Course” Deserae Cain, 33, is one of the 4,000 patients still relying on a HeartWare device. She was implanted with the heart pump in late 2017, after suddenly being diagnosed with heart failure. Scans showed her heart was three times normal size. It took time for her to come to terms with needing a life-sustaining device — not long before her diagnosis, she had been going on five-mile runs. In the four years since, though, Cain has built a life around the HVAD with her fiance in their Dayton, Ohio, home. They know the device can malfunction. In 2019, the pump failed for almost an hour as doctors at a nearby hospital struggled to restart it. Cain just tried to stay calm, knowing anxiety could threaten her unsupported weak heart. Months later, she needed an emergency experimental procedure to clear out blood clots developed within her HVAD. Then, in 2020, Cain developed a widespread infection. Doctors told her she needed surgery to clean out and replace the pump. Cain asked her medical team if she could switch to the alternative HeartMate device, which other patients told her presented fewer problems, she said. Doctors said the HVAD was better suited for her smaller frame. But her new pump had problems soon after the surgery. The device’s suction alarms, which alert when the pump is trying to pull in more blood than is available within the heart, sounded multiple times a day, for hours at a time, she said. Baffled by the issue for months, her medical team eventually turned off that specific alarm. Soon after, her ventricular-assist specialist called her about a patient’s death linked to the belt that holds the device controller, she said. The belt had ripped and the equipment had fallen, yanking on the cable that connected the controller to the pump. Cain replaced her belt but it quickly frayed and had to be replaced again within six weeks. Then, in June, she found out about Medtronic’s decision to stop sales and implants. Cain received a letter from her hospital mentioning a Medtronic support program, but it provided few specifics. Cain wondered if things would be any different than before. Anxious about her future, she asked: “Are they just going to let it run its course until there is none of us left?” Source URL |